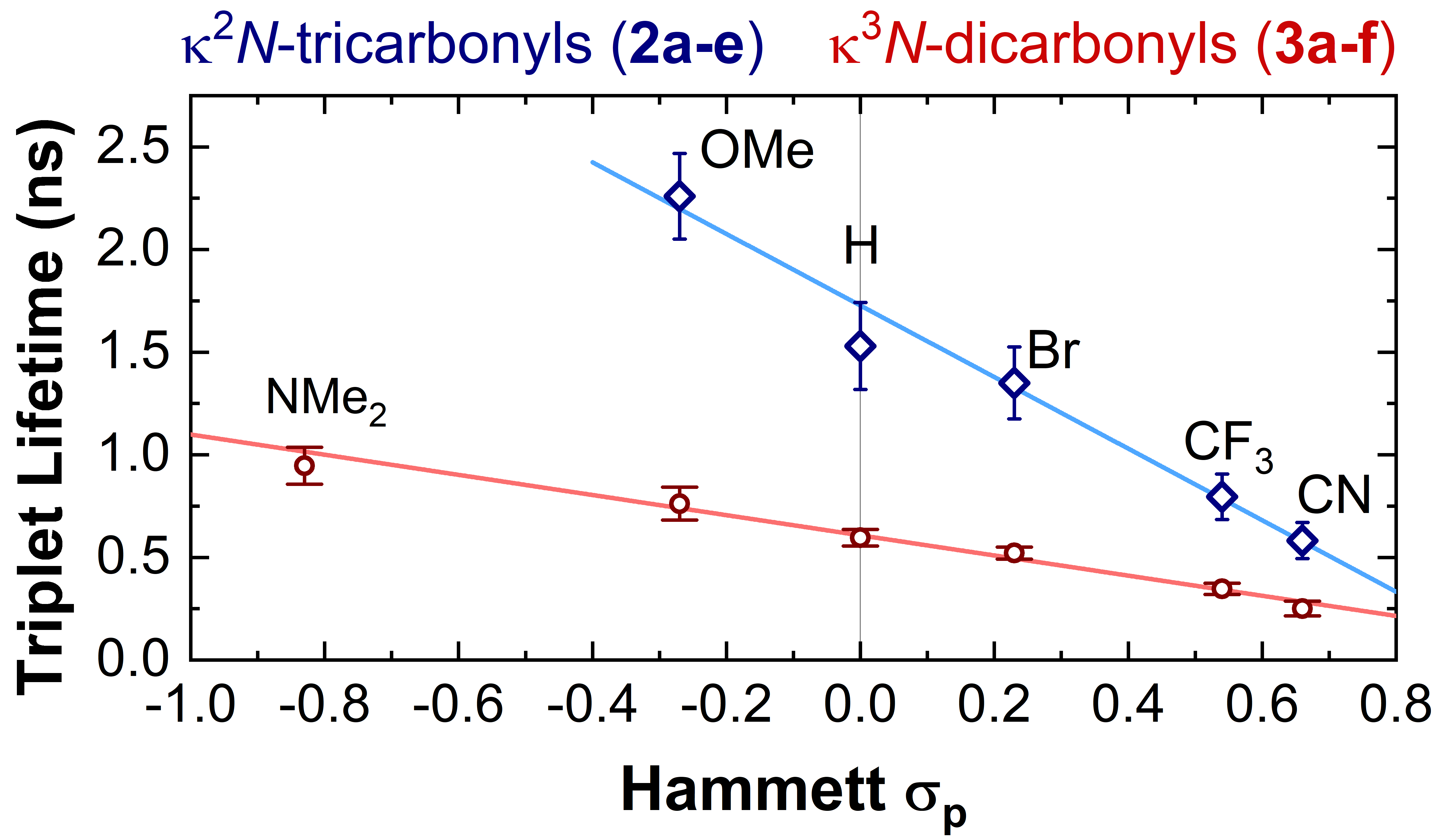
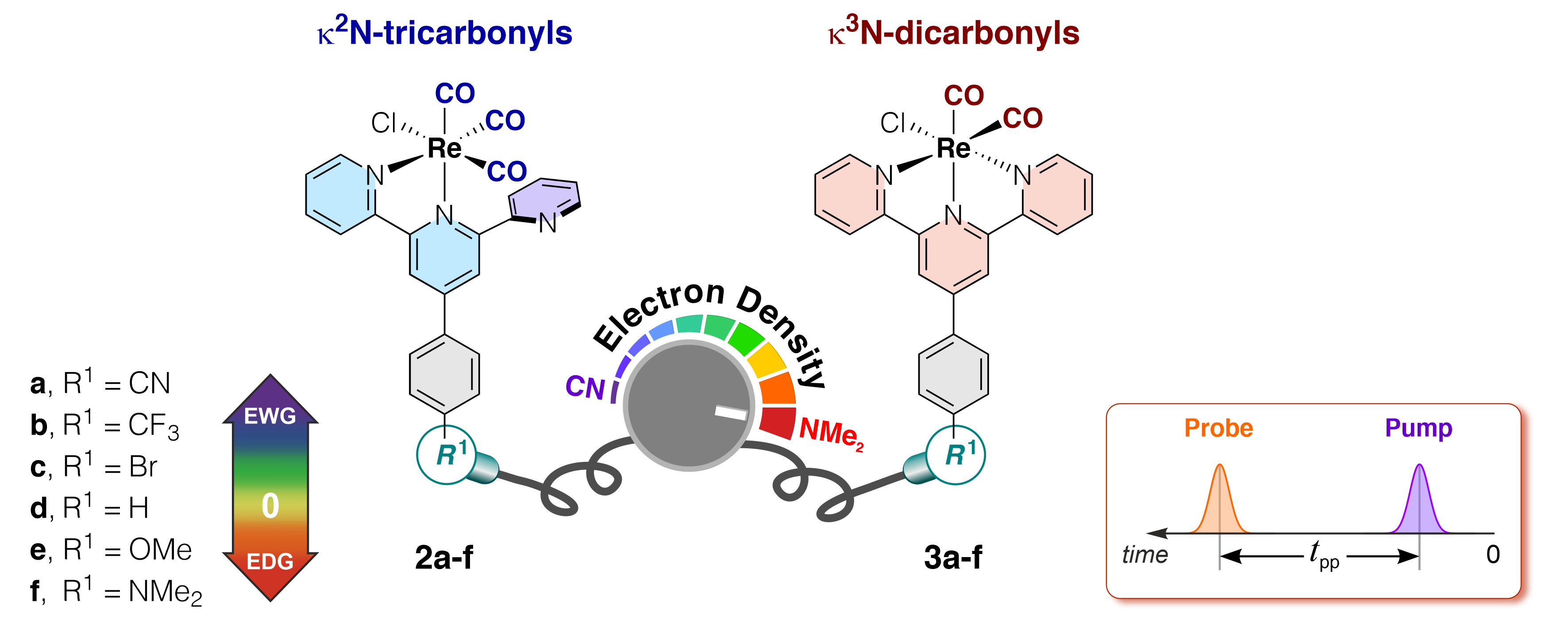
- A systematic modification in the triplet lifetimes was observed: more electron-donating substituents increase the lifetime.
- The ground-state electrochemical potentials of the k2N-tricarbonyl complexes are also systematically changed: complexes with electron-donating substituents are stronger (photo)reductants.
- The complexes 2a-e are emissive and show a strong rigidochromic blue shift upon cooling to 77 K (in 2-MeTHF), characteristic of 3MLCT excited states.
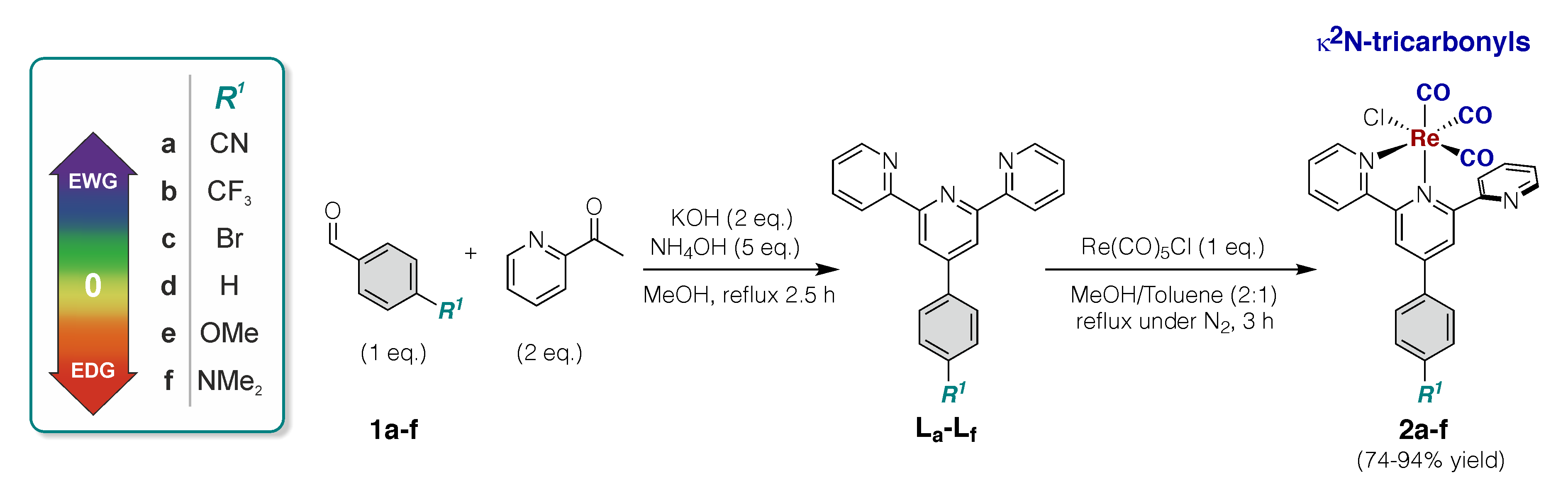
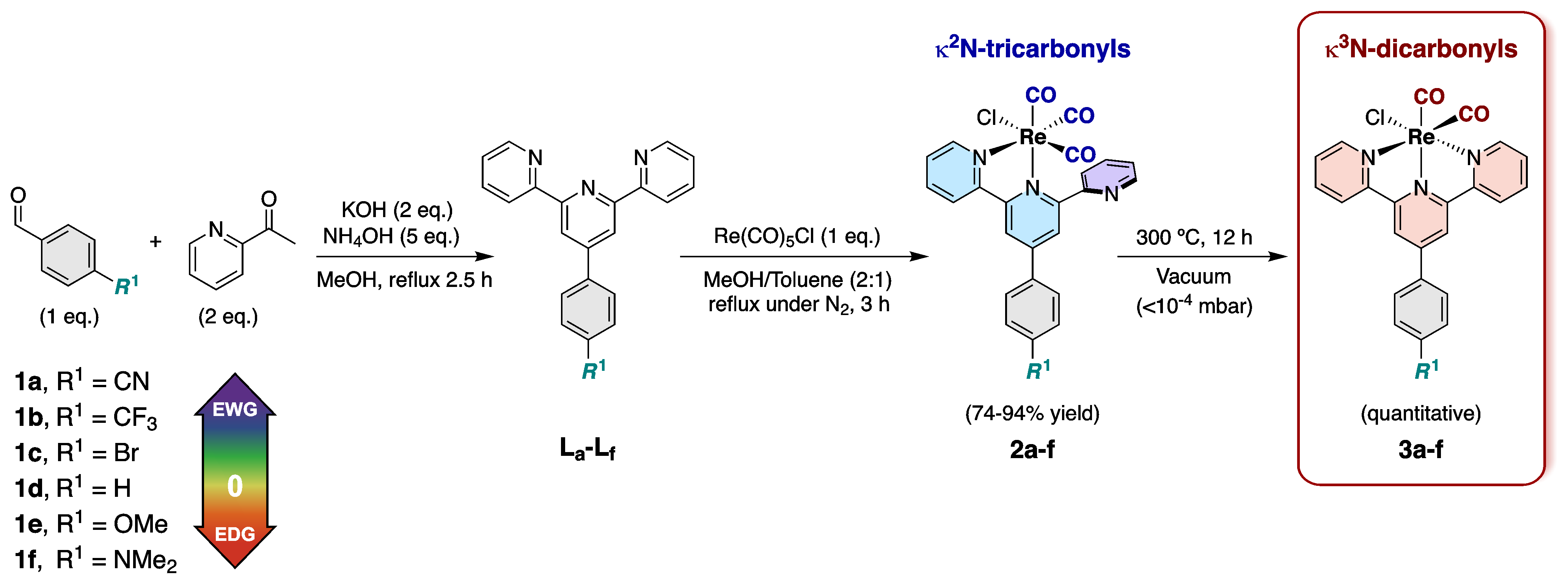
Synthetic route and structures of the studied complexes
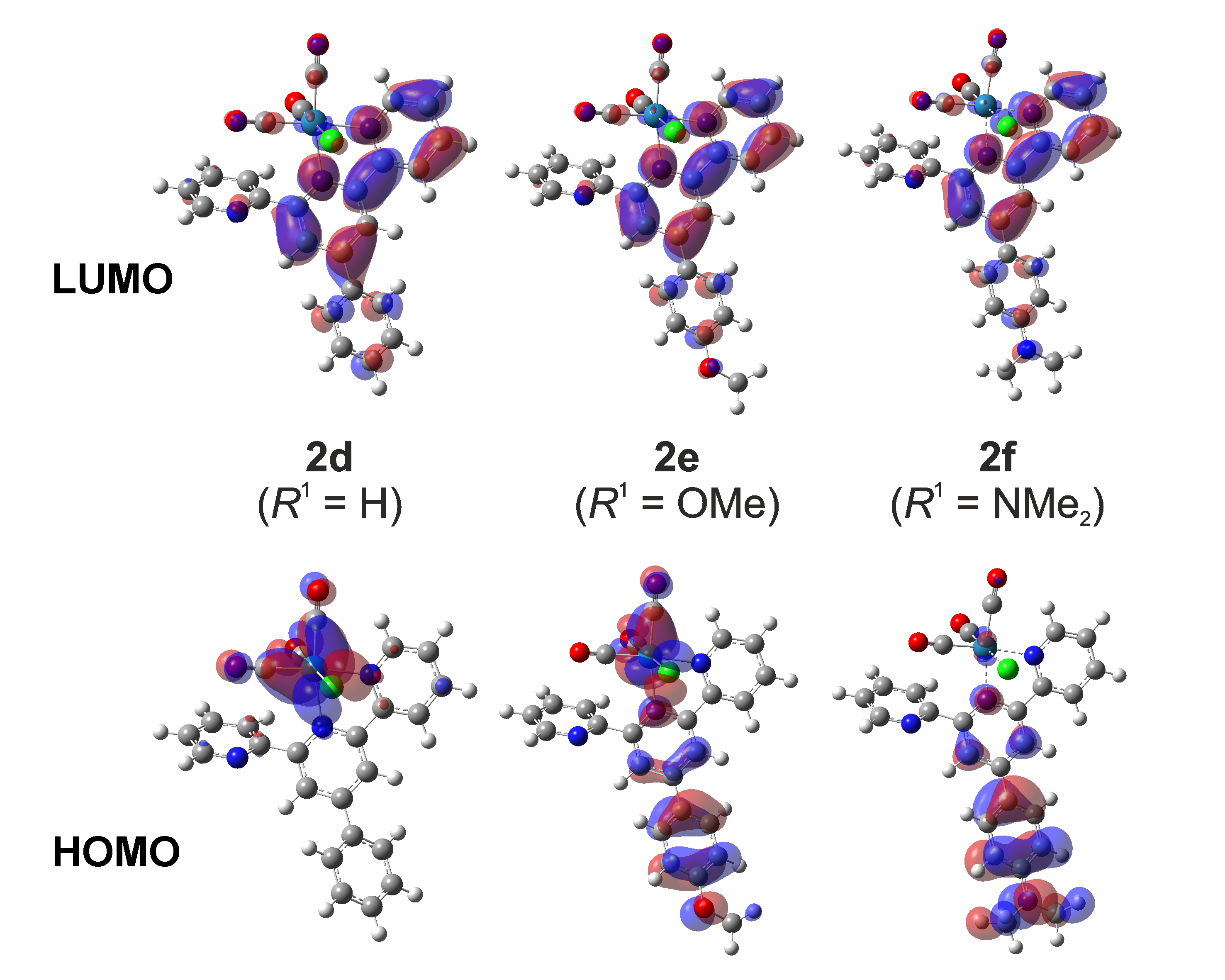
HOMO and LUMO orbitals of the complexes with more electron-donating substituents (2e-f) and the unsubstituted/reference complex (2d)
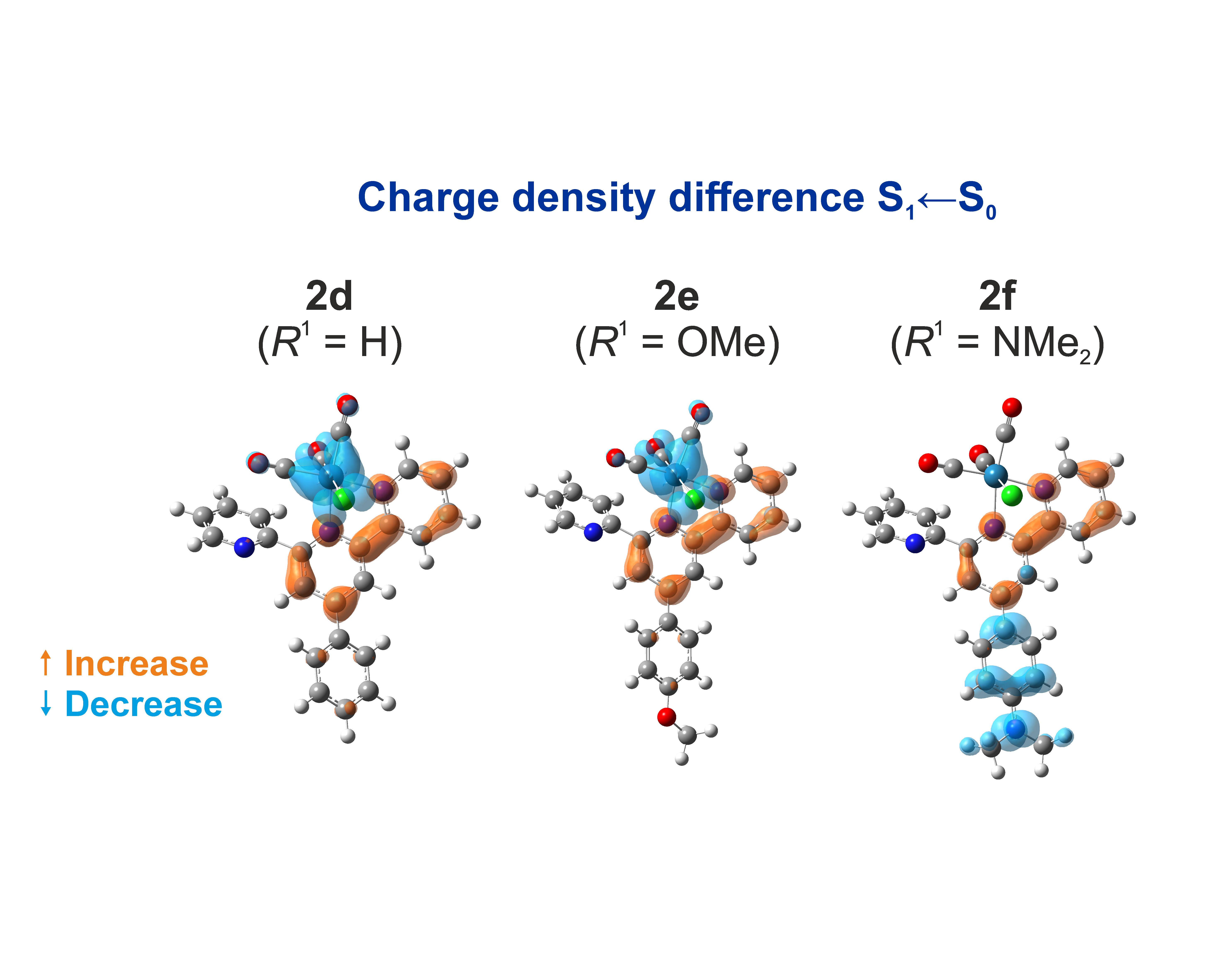
Charge density difference isosurfaces of the S1 excitation of the complexes with more electron-donating substituents (2e-f) and the unsubstituted/reference complex (2d)
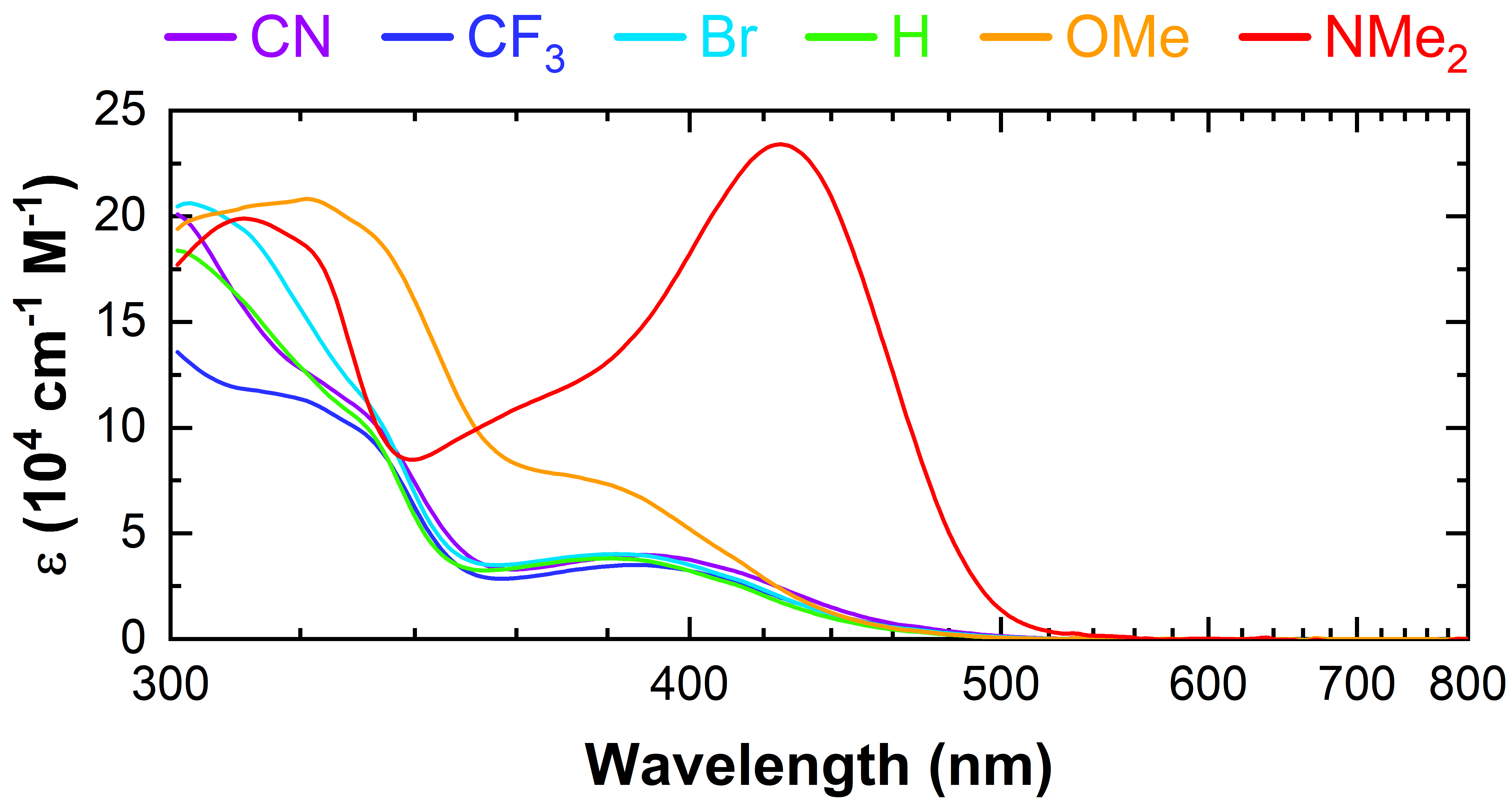
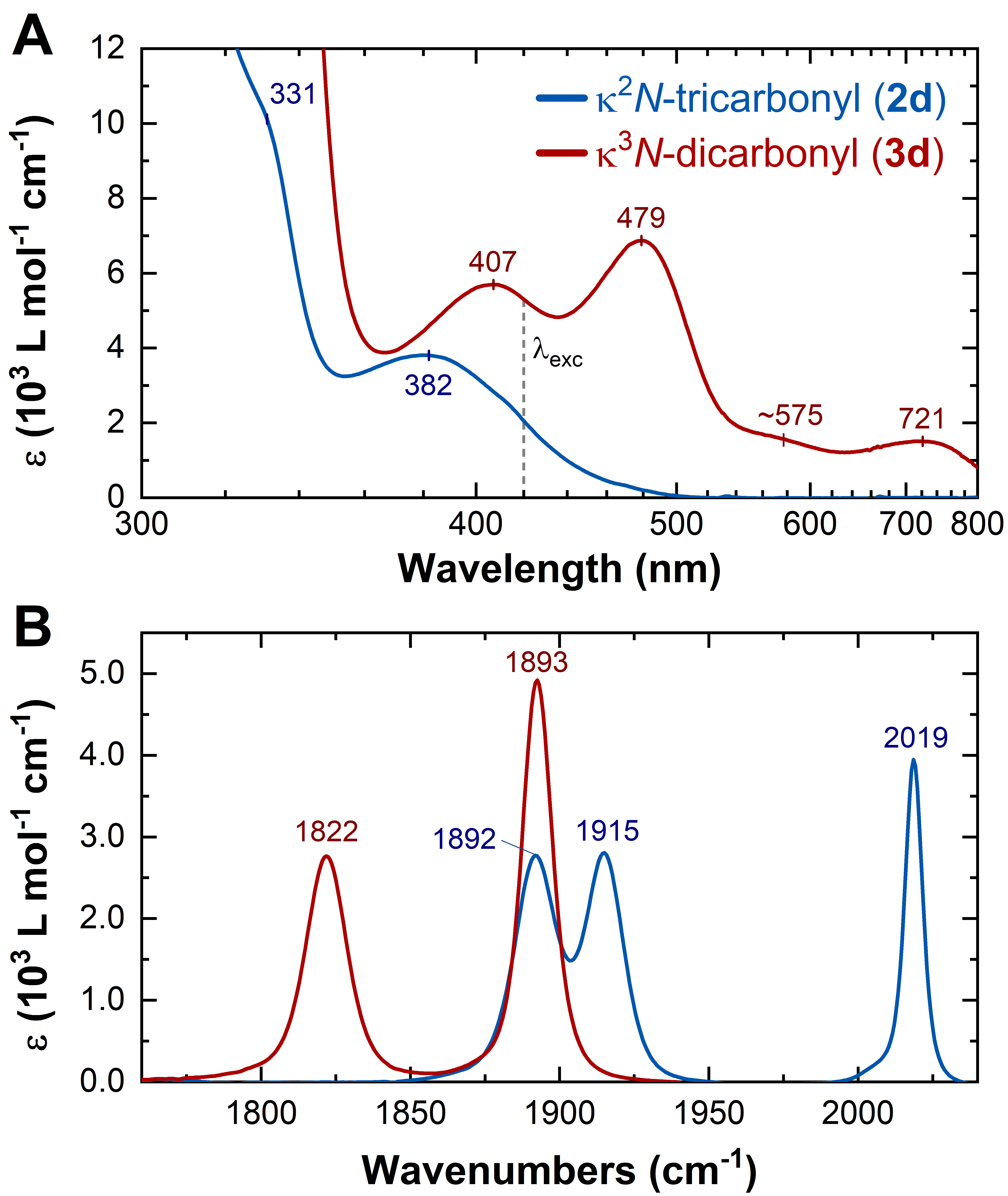
We evidenced a systematic shift in the UV-Vis absorption spectra, where an additional very intense transition was observed in the case of the NMe2-substituted complex (2f).
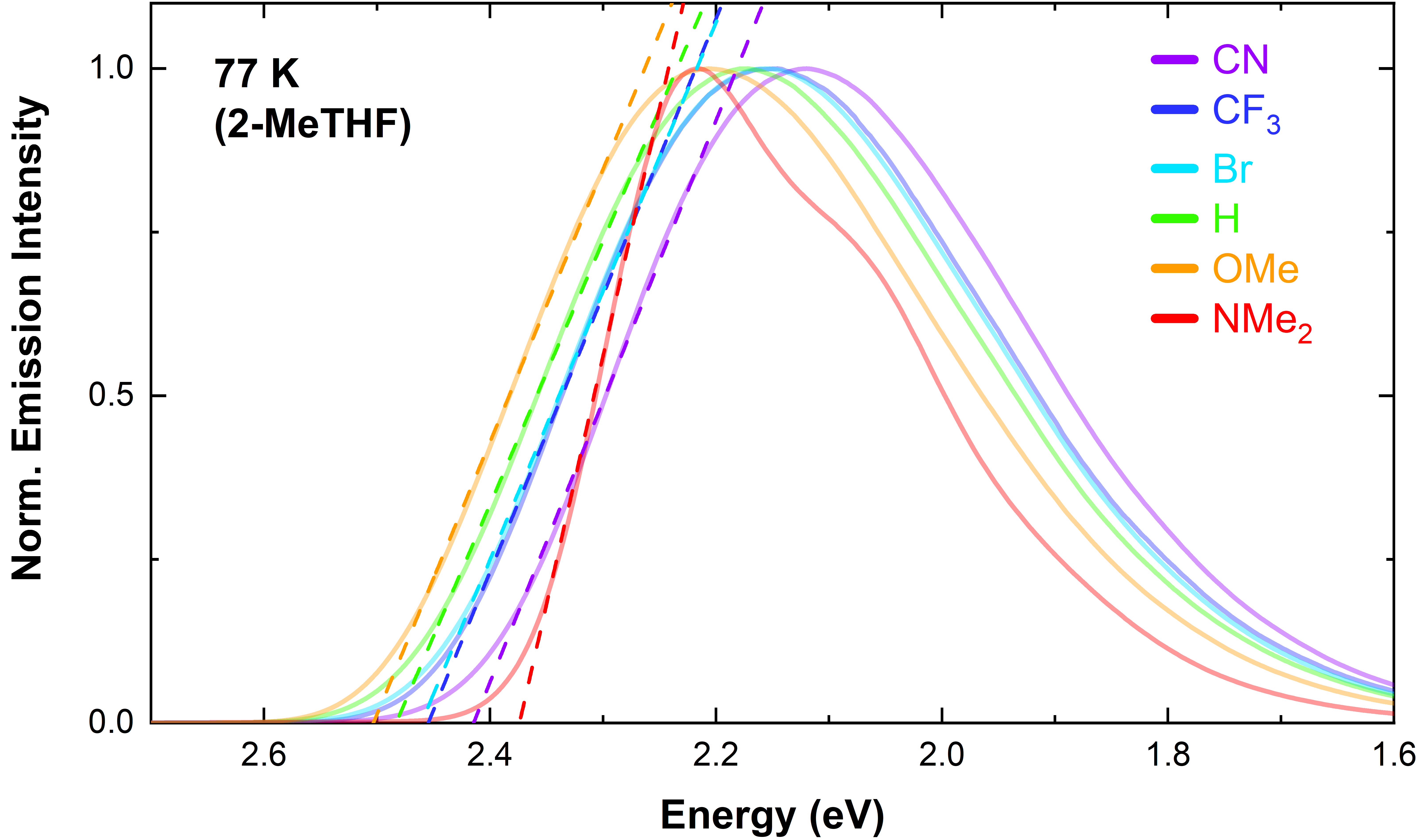
Low-temperature emission spectra allowed us to determine the singlet-triplet energy gap ΔGST, crucial to obtaining the excited-state redox potentials.
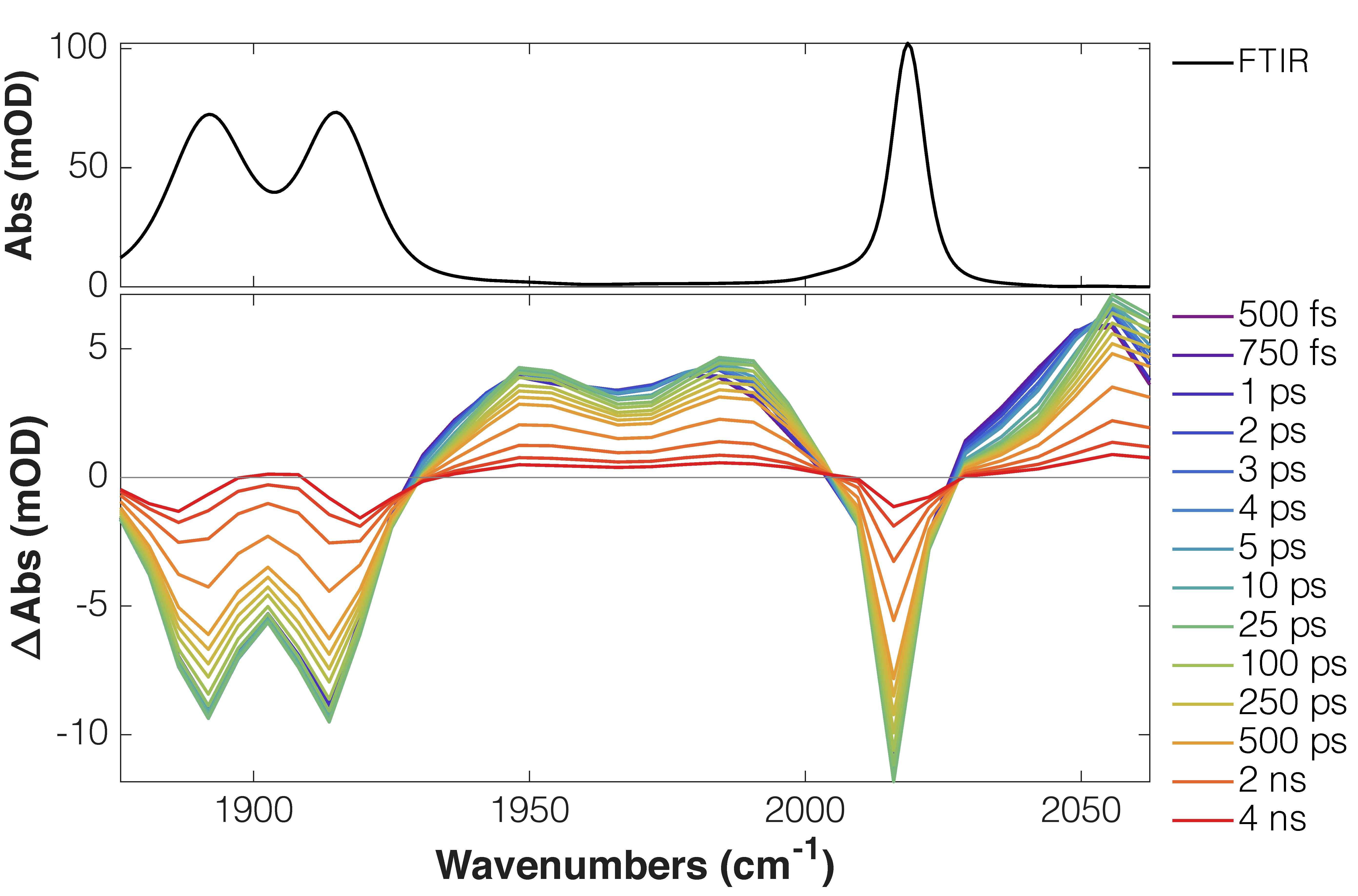
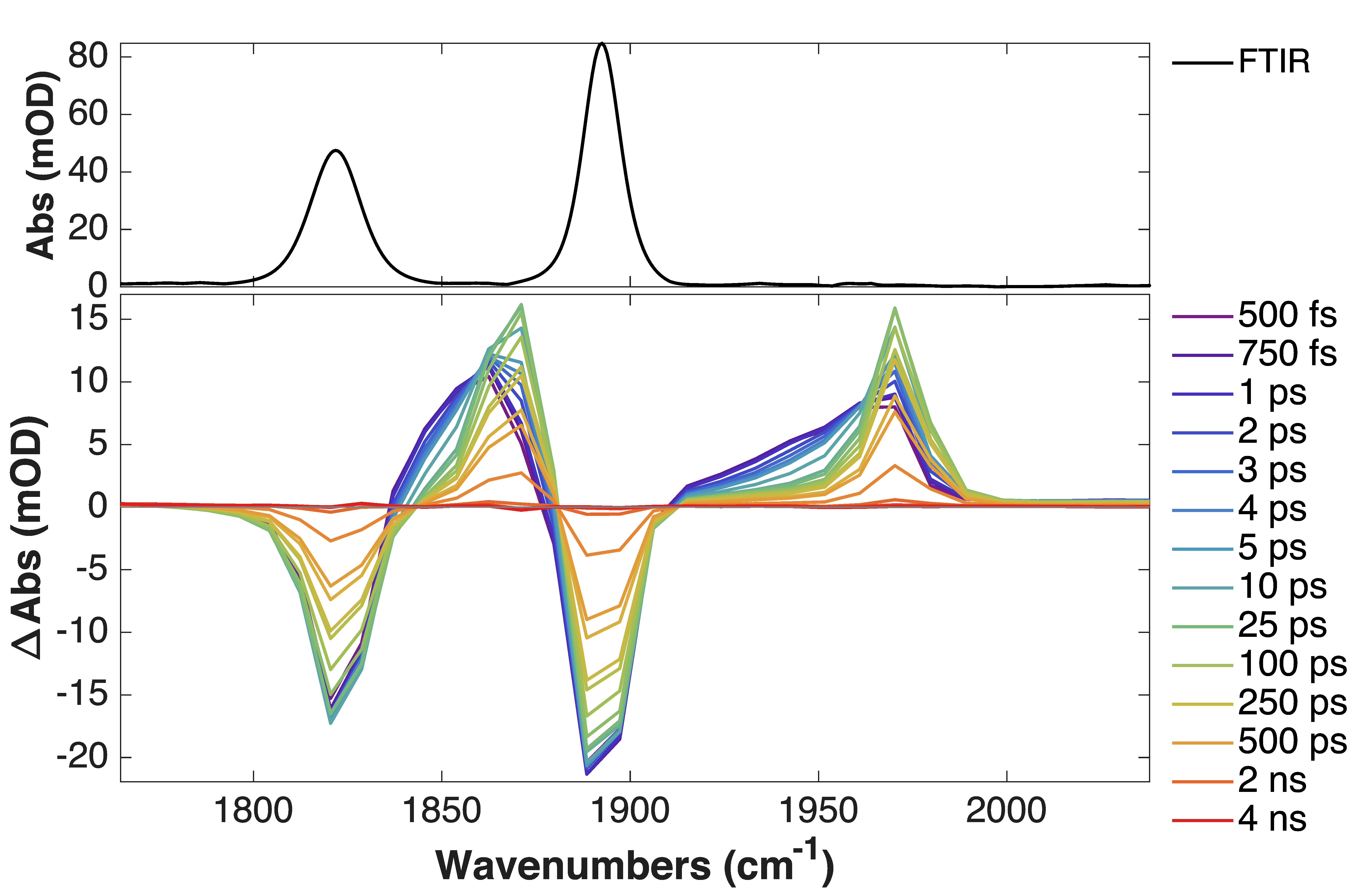
TRIR spectra of complexes 2a-e (R1= CN to OMe) exhibited the typical features of an MLCT excited state (i.e. blue-shifted excited-state absorption bands)
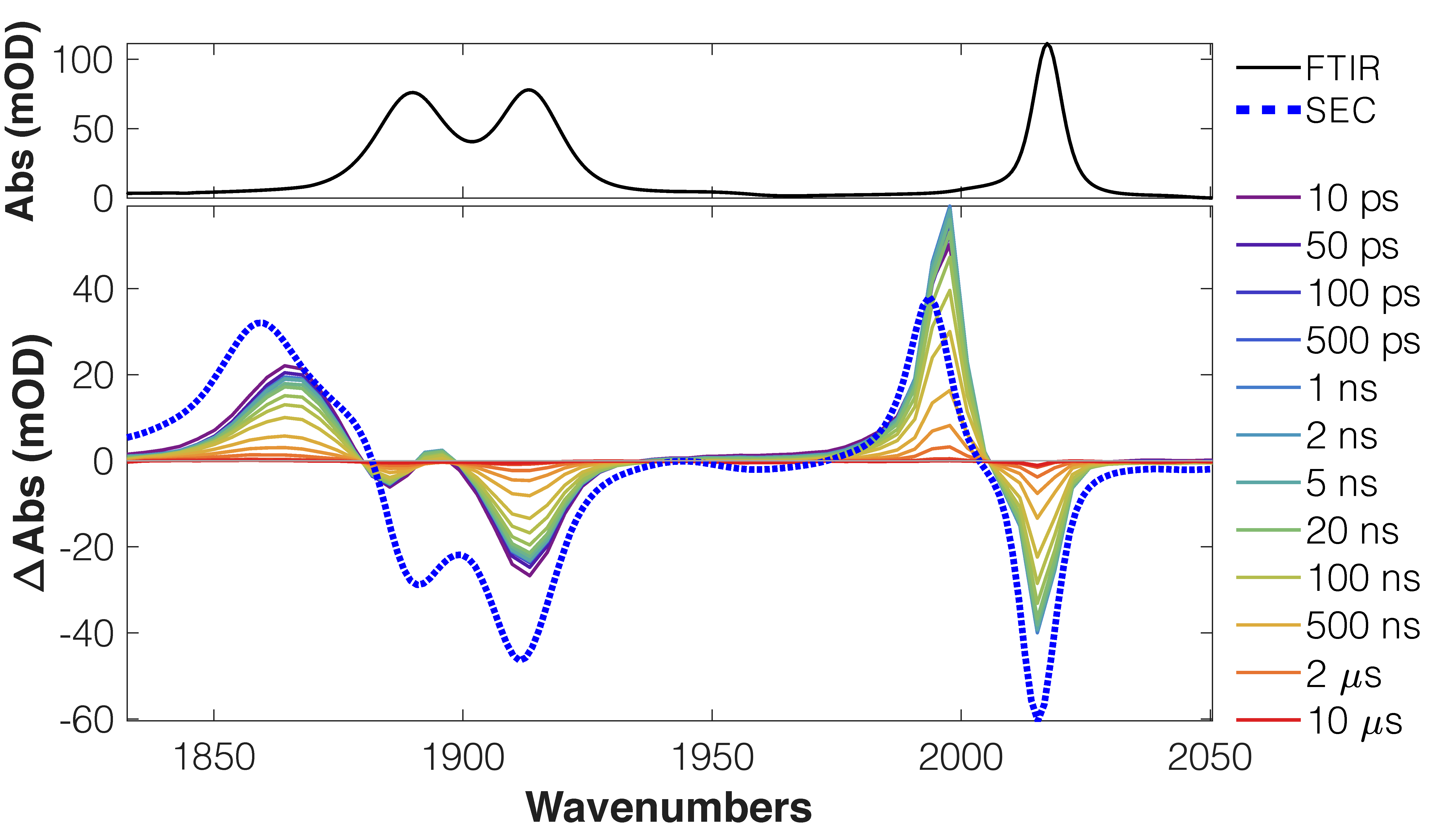
TRIR spectra of complex 2f (R1 = NMe2) shows features attributed to an intraligand charge transfer (ILCT) state, which compare favourably with the difference spectrum of the 1st reduction (IR-SEC, blue dashed line).
A significantly longer lifetime (380 ns for 2f, vs 2 ns for the unsubstituted complex, 2d) was observed in this case.
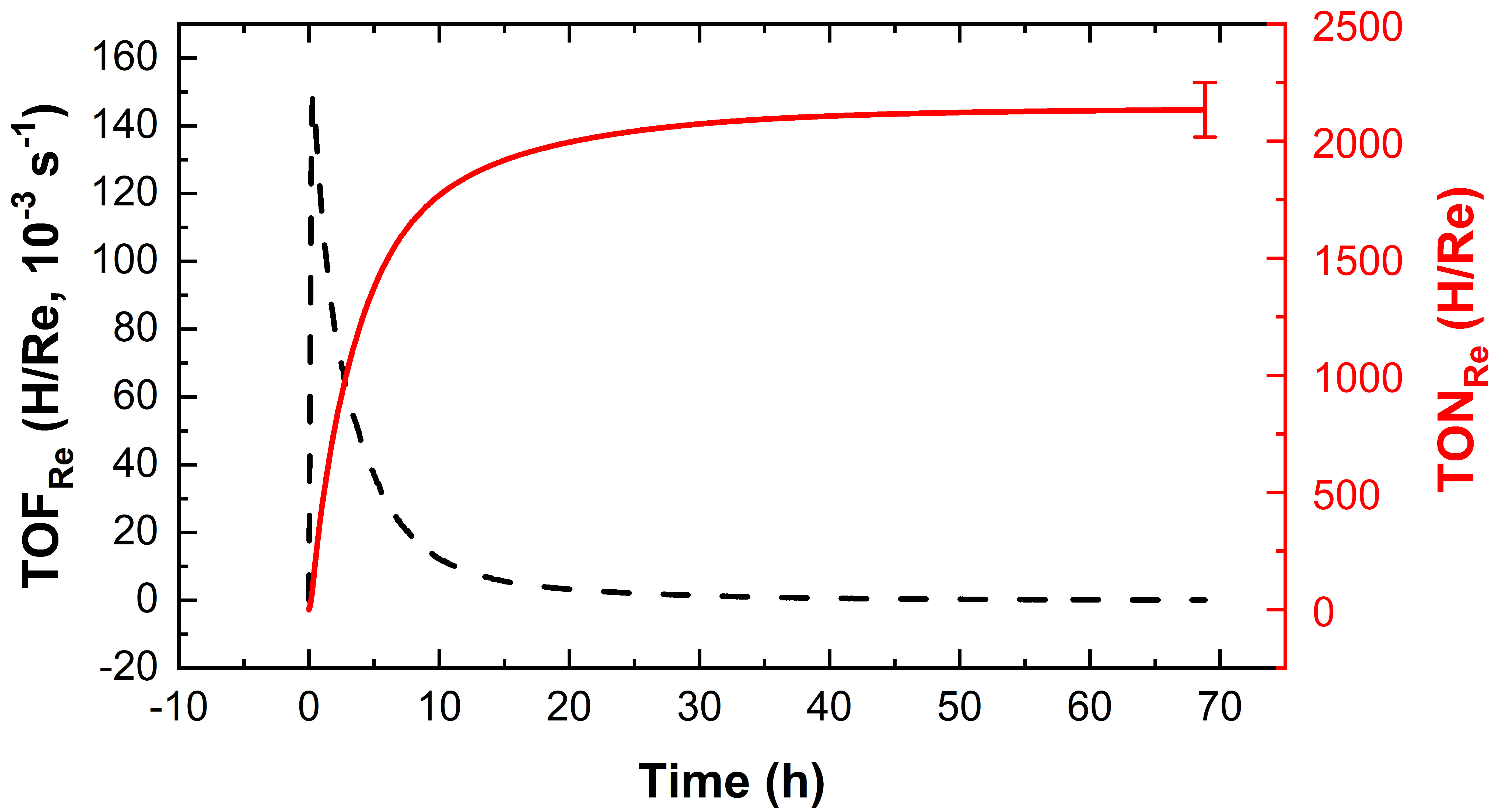
Hydrogen evolution from 2f as a photosensitizer. Conditions: [2f] = 50 μM, [Co] = 0.5 mM, [dmgH2] = 3.5 mM, 1
M TEOA, 0.1 M TfOH in DMF, and λexc = 453 nm.
Sustained hydrogen evolution was observed when combining complex 2f with a standard Cobalt-DMG catalyst in DMF with triethanolamine (TEOA) as sacrificial electron donor.
The initial electron transfer step in this photocatalytic system is believed to consist of oxidative quenching of the [Re]* photosensitiser, followed by very quick regeneration by TEOA.
This shows that ILCT states can be used to harvest more sunlight through a ca. 100 nm red shift of the absorption, while mantaining similar redox potentials and longer lifetimes than MLCT states in the same series of complexes.